Síndrome de Dravet: sintomas, causas, tratamentos
A síndrome de Dravet é um tipo de criança que apresenta epilepsia caracterizada por resistência ao tratamento e evolução clínica para outros tipos de crises epilépticas e comprometimento cognitivo grave (Sánchez-Carpinterio, Núñez, Aznárez e Narbona García, 2012).
No nível etiológico, a síndrome de Dravet é uma doença de origem genética associada a mais de 500 mutações diferentes, no entanto, aproximadamente 70% dos afetados apresentam uma alteração específica no gene SCN1A, localizado no cromossomo 2 (Mingarro Castillo, 1998). Carmona de la Morena, Latre Martínes e Aras Portilla, 2014).

Em relação ao curso clínico, esse distúrbio é caracterizado pelo desenvolvimento de crises epilépticas recorrentes durante o primeiro ano de vida. Geralmente, as crises são de natureza mioclônica generalizada e geralmente são acompanhadas por episódios febris (Jiang, Shen, Yu, Jiang, Xu, Xu, Yu, Gao, 2016).
Além disso, a síndrome de Dravet é considerada um dos tipos mais graves de epilepsia, uma vez que quase todas as crianças afetadas progridem para deterioração neurológica grave ou muito grave (Nieto-Barrera, Candao e Nieto-Jiménez, 2003).
O diagnóstico desse tipo de patologia é semelhante ao de outras epilepsias, com base no exame clínico, nas características dos episódios convulsivos e no uso de exames laboratoriais como o eletroencefalograma.
Além disso, uma cura para a síndrome de Dravet ainda não foi identificada, é um tipo de epilepsia que resiste ao tratamento medicamentoso, no entanto, a combinação de diferentes abordagens médicas pode retardar sua progressão (Mingarro Castillo et al., 2016).
Características da síndrome de Dravet
A síndrome de Dravet, também conhecida como epilepsia mioclônica grave na infância (IMGI), é definida como uma forma catastrófica e infrequente de epilepsia resistente ao tratamento que tem início típico na fase da infância (Dravet Syndrome Foundation, 2016).
Como sabemos, a epilepsia é um dos distúrbios neurológicos mais freqüentes do curso cônico na população em geral. Assim, esta patologia é caracterizada pela presença recorrente de convulsões (Epilepsy Foundation, 2016).
Na epilepsia, os padrões habituais e funcionais da atividade elétrica cerebral estão alterados, resultando no desenvolvimento de espasmos musculares, perda de consciência, alterações de comportamento ou percepção de sensações estranhas (Instituto Nacional de Distúrbios Neurológicos e Derrames, 2016). .
Embora existam vários tipos de epilepsias e tipos de convulsões, a síndrome de Dravet é classificada como epilepsias mioclônicas, caracterizadas por crises ou episódios de contração muscular bilateral (Asociación Andaluza de Epilepsia, 2016).
Especificamente, essa síndrome foi inicialmente identificada por Charlotte Dravet em 1978 (Pérez e Moreno, 2015). Em seu relato clínico, ele se referiu à identificação de vários casos de epilepsia grave, resistente ao tratamento medicamentoso e com algumas características clínicas compartilhadas com a síndrome de Lennox-Gastaut (Nieto-Barrera, Candao e Nieto-Jiménez, 2003).
Paralelamente a Dravet, outros pesquisadores, como Scheffer e Bervic, descrevem uma síndrome epiléptica de origem genética caracterizada pela presença de convulsões febris, na qual a síndrome de Dravet é considerada o fenótipo mais comum (Pérez e Moreno, 2015).
No entanto, não foi até 1985, quando a Liga Internacional de Epilepsia incluiu-o dentro das "Epilepsias e síndromes indeterminadas em relação à localização com a apresentação de crises generalizadas e focais" (Pérez e Moreno, 2015).
Além disso, as pesquisas mais recentes indicam que a síndrome de Dravet é uma condição médica realmente séria e debilitante que, para além da familiar, prejudica significativamente a qualidade de vida da pessoa afetada (Dravet Fundação Síndrome, 2016).
Além dos sinais e sintomas caracterizados por episódios convulsivos, essa síndrome tende a evoluir para a presença de atrasos significativos no desenvolvimento, distúrbios comportamentais, déficits cognitivos, etc. Além disso, apresenta alta comorbidade com outros tipos de condições médicas, como a morte súbita (Dravet Syndrome Foundation, 2016).
Estatísticas
Estudos epidemiológicos indicam que a síndrome de Dravet tem uma incidência de aproximadamente 1 caso por 20.000.40.000 nascimentos. No entanto, protocolos diagnósticos e novos procedimentos médicos podem aumentar significativamente esse número, uma vez que permitem progressivamente um diagnóstico cada vez mais precoce (Dravet Syndrome UK, 2016).
Além disso, sua prevalência é estimada em cerca de 7% dos tipos de epilepsia que aparecem durante a fase precoce da infância, ou seja, aqueles com menos de três anos de idade (Mingarro Castillo et al., 2014).
Em relação à sua distribuição demográfica, a síndrome de Dravet afeta homens e mulheres de maneira semelhante, e uma maior prevalência associada a regiões geográficas específicas e / ou grupos étnicos ou raciais específicos não foi identificada (Mingarro Castillo et al., 2014).
Por outro lado, se nos referirmos aos dados relacionados ao seu curso clínico, entre 3-7% das pessoas afetadas têm os primeiros episódios convulsivos antes de atingirem a idade de um ano, enquanto 7% geralmente o desenvolvem durante os 3 anos ( Pérez e Moreno, 2015).
Além disso, em famílias afetadas por diferentes casos de epilepsia, a síndrome de Dravet geralmente ocorre em um de seus membros em mais de 25% do tempo (Pérez e Moreno, 2015).
Signos e sintomas
Como apontamos, os achados médicos fundamentais na síndrome de Dravet são convulsões de natureza epiléptica e convulsões febris:
a) Convulsões
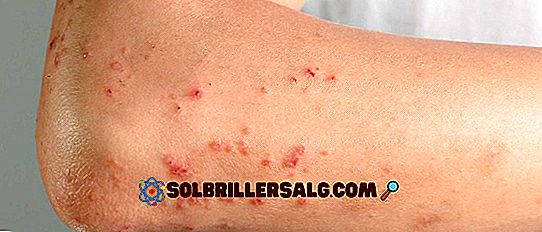
Episódios convulsivos desenvolvem-se como resultado de atividade neuronal anormal, generalizada ou focal. Eles geralmente causam espasmos musculares e / ou perda de consciência, embora existam tipos diferentes. No caso da síndrome de Dravet, os mais comuns são (Instituto Nacional de Distúrbios Neuropáticos e Derrames, 2015):
- Crise tônica : neste caso, os episódios são caracterizados pelo desenvolvimento de tensão muscular, tendendo à rigidez generalizada, principalmente nas pernas e nos braços.
- crises mioclônicas : neste caso, os episódios são caracterizados pelo desenvolvimento de espasmos musculares repetitivos e descontrolados, em um nível visual, eles podem ser observados como agitação do corpo.
- Crise tônico-clônica : neste caso, os episódios são caracterizados por uma combinação dos anteriores, além disso, eles tendem a apresentar em paralelo uma perda temporária de consciência.
b) Crise febril
Juntamente com as crises descritas na seção anterior, é comum observar o desenvolvimento de episódios de febre alta, ou seja, aumento anormal da temperatura corporal, geralmente acima de 37 graus.
Assim, diferentes autores chamam essa condição médica de convulsão febril, pode fazer parte de uma patologia epiléptica ou aparecer no quadro outras doenças que ocorrem com febre alta.
Além desses sintomas cardinais, a síndrome de Dravet apresenta um curso clínico específico que descreveremos a seguir (Dravet Syndrome UK, 2016, López, Varela e Marca, 2013, Sánchez-Campiertero, Núñez, Aznárez e Narbona Garcia, 2012):
Os primeiros sinais e sintomas geralmente aparecem antes do primeiro ano de vida, ou seja, no estágio neonatal ou de lactação. Assim, convulsões geralmente aparecem junto com episódios febris, tendem a ser tônico-clônicas generalizadas e afetam o corpo bilateralmente ou unilateralmente.
Além disso, as primeiras crises geralmente têm uma duração prolongada, de mais de 5 minutos, de modo que geralmente requerem intervenção médica urgente. Por outro lado, nos primeiros momentos do desenvolvimento da síndrome de Dravet, o desenvolvimento neurológico geralmente não é afetado inicialmente.
À medida que esta doença progride, após vários meses de sua apresentação, as crises geralmente se tornam mais frequentes e intensas, no entanto, os episódios de febre alta têm que desaparecer. Nesse caso, as crises geralmente são mioclônicas, embora possa haver algumas crises de ausência, caracterizadas pela falta de resposta do indivíduo.
Por outro lado, quando a pessoa atinge o estágio entre 2 e 4 anos de idade, as crises epilépticas prolongadas se tornam um sério risco para a integridade neurológica.
Nessa fase, já é possível identificar atrasos significativos no desenvolvimento, déficits cognitivos e outros sintomas, como ataxia, distúrbios do sono ou distúrbios de comportamento.
Assim, a evolução usual da síndrome de Dravet é direcionada para uma epilepsia resistente ao tratamento farmacológico, com retardo e grave estagnação do desenvolvimento psicomotor e o sofrimento de diferentes déficits cognitivos em um espectro de moderado a grave.
Normalmente, com o passar do tempo, identificou-se uma tendência à estabilização, que permite o desenvolvimento de diversas capacidades, como a linguagem ou o andar funcional.
Causas
A síndrome de Dravet é uma epilepsia de origem genética, embora possa estar associada a uma ampla variedade de anomalias genéticas, especificamente mais de 500 mutações específicas, a maioria das quais está associada a mutações no gene SCN1A (National Organization Para desordens raras, 2016).
Em um nível específico, esse gene está localizado no cromossomo 2 e é responsável pela codificação de uma subunidade alfa 1 de canais de sódio dependentes de voltagem, na qual seu funcionamento eficiente é fundamental para a transmissão correta dos impulsos nervosos entre os neurônios. (Mingarro Castillo et al., 2014).
Embora as formas específicas da mutação genética geralmente não se correlacionem completamente com o espectro clínico específico, é mais provável que a pessoa afetada apresente uma sintomatologia mais grave se parecer uma mutação de novo (aleatória), que se a doença for produto de uma transferência hereditária (National Organization For Rare Disorders, 2016).
Além disso, mutações específicas no gene SCN1A também estão relacionadas a outros cursos epilépticos, incluindo:
- Epilepsia generalizada com convulsões febris.
- Epilepsia multifocal infantil grave.
- Derrame epiléptico infantil esporádico.
- Crises de infância tônico-clônicas intratáveis.
Diagnóstico
Em geral, os diferentes subtipos de epilepsia são diagnosticados com base nas características clínicas dos episódios convulsivos: é essencial conhecer o tempo da apresentação inicial, a frequência, a duração e a forma das convulsões.
Além disso, outro aspecto importante é o exame físico e neurológico, para identificar outros tipos de sintomas que acompanham a crise, como febre, comprometimento cognitivo, entre outros.
Por outro lado, outra medida básica é o estudo eletroencefalográfico, pois nos oferecerá informações sobre a organização da atividade cerebral da pessoa afetada.
Além disso, na síndrome de Dravet, um estudo genético é realizado para identificar anomalias compatíveis com mutações genéticas e outros tipos de alterações.
Tratamento
A síndrome de Dravet é um tipo de epilepsia resistente aos medicamentos, ou seja, resistente a todas as abordagens farmacológicas clássicas ou combinadas (Pérez e Moreno, 2015).
No entanto, diferentes foram concebidos com base na combinação de alguns medicamentos, como valproato, clobazan ou sitirioentol, que são capazes de retardar a progressão das crises. No entanto, eles têm efeitos adversos significativos (Pérez e Moreno, 2015).
Além disso, outros tipos de drogas também são usados para reduzir a intensidade e a duração dos episódios, como clonazepam, leviteracetam, ácido valpróico ou tipiramato (Dravet Syndrome Foundation, 2016).
Por outro lado, também foram identificadas algumas abordagens que tendem a piorar o quadro clínico, tais como: cabarmazepina, fosfenitoína, lamotrigina, oxcarbazepina, fenitoína e vigabatrina (Dravet Syndrome Foundation, 2016).